CHILD AND ADOLESCENT PSYCHIATRIC CLINICS
Child Adolesc Psychiatric Clin N Am OF NORTH AMERICA
13 (2004) 623—640
Androgen insensitivity syndrome and
Klinefelter’s syndrome: sex and
gender considerations
Milton Diamond, PhDa,*,
Linda Ann Watson, MEdb
aDepartment of Anatomy and Reproductive
Biology, John A. Burns School of Medicine,
University of Hawaii, Manoa, 1960 East-West Road, Honolulu, HI 96822, USA
bDepartment of Counseling and Guidance, University of Hawaii, Manoa, Honolulu,
HI 96822, USA
The
androgen insensitivity syndrome (AIS) and Klinefelter’s syndrome (KS), which
usually are the province of endocrinologists and geneticists, present features
of importance to psychiatrists and other psychotherapists. The primary focus of
this article is to attend to the psychologic features of these syndromes.
Although not common, these conditions are not rare. They are among the
most commonly seen intersex conditions and have a prevalence of 2 or 3 cases/1
000 population. These conditions present instances of undermasculinization and
both syndromes can occur in the same individual [1].
The medical-clinical-molecular characteristics of these conditions are covered
in detail elsewhere [2—6]. We start with a brief introduction to the medical
nature of each of these syndromes before proceeding to psychologic and social
considerations.
Androgen
insensitivity syndrome
A result of potentially
hundreds of genetic mutations to the androgen receptor gene [7], the AIS is manifest
by a notable inability of an individual who has XY sex chromosomes to respond
to androgens. This inability occurs despite the presence of testes and typical
testosterone production, transport, and metabolism [8]. Of particular
consequence is the relative or complete failure of the individual to respond to
testosterone or dihydrotestosterone that is crucial for the organi-
__________
Support for this
work has come from the Eugene Garfield Foundation, Philadelphia, Pennsylvania.
* Corresponding author.
E-mail address: diamond@hawaii.edu (M. Diamond).
1 056-4993/04/$ — see front matter © 2004 Elsevier Inc. All rights
reserved.
doi: 10. 1016/j.chc.2004.02.015
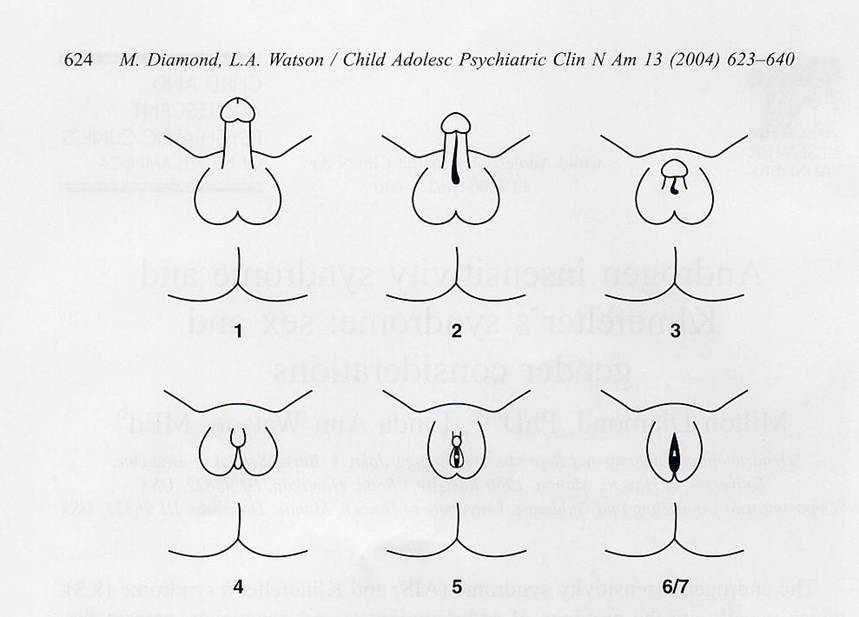
Fig. 1. Schematic
representation of a grading scheme for clinical classification of AIS. Grades
are numbered I through 7 in order of increasing severity (more defective
masculinization). Grade 1:
normal masculinization in utero; grade 2: male phenotype with mild defect in
masculinization (eg, isolated hypospadias); grade 3: male phenotype with severe
defect in masculinization—small penis, perineoscrotal hypospadias, bifid
scrotum or cryptorchidism; grade 4: severe genital ambiguity— clitoral-like
phallus, labioscrotal folds, single perineal orifice; grade 5: female
phenotype with posterior labial fusion and clitoromegaly; grade 6/7: female
phenotype (grade 6 if pubic hair present in adulthood, grade 7 if no pubic hair
in adulthood). (A dapted from Quigley CA, DeBellis A, Marschke KB, El-Awady MK,
Wilson EM, French FS. Androgen receptor defects: historical, clinical, and
molecular perspectives. Endocrine Rev 1995; 1 6(3):282; with permission.)
zation and
activation of the anatomic and neural features that are needed for typical male
development [5,9,10].
Persons who have AIS have genitalia that can look typically female
in appearance (complete AIS; CAIS) or ambiguous with features that range from
typical male-like to typical female-like (partial AIS; PAIS) (Fig. 1). Tissue
response to estrogen is present and breast development and other signs of
feminization occur. Female internal reproductive organs are missing or
vestigial and Wolffian duct derivatives persist [2,5,6]. No relation
between gene mutation and phenotype has been discovered. Infertility is common.
Persons who have CAIS appear female at birth and are reared as girls.
Unless there is known or suspected AIS in the family or inguinal testes are
detected, these girls typically go undiagnosed until puberty. The vagina may be
short and blind-ending and may or may not need elongation [11]. Breast
development occurs but pubic and axillary hair development is sparse or absent.
When menses fails to occur a remedy is sought. Those who have PAIS usually are
noticed at, or soon after, birth. Depending upon the
degree of masculinization of the genitals, the child may be raised as a boy or
a girl.
AIS is an inherited condition that is propagated as a recessive X-linked
single gene syndrome that can manifest differently in children of the same
parents; one child in a family can be raised as a boy, whereas another can be
raised as a girl [12].
A study of individuals in the United Kingdom, who had AIS and were 16
years of age or younger, found that 59% of those who were identified as
having PAIS were assigned as males [13]. Most published cases of
clinical-psychiatric involvement, however, seem to be related to subjects who
were raised as females.
In contrast with, and usually of greater impact on the person than any
medical morbidity, are the psychologic concomitants to the syndrome and
management of related psychological problems. Concerns vary, depending upon
whether the individual has the complete or partial form of the condition, if
the individual was raised as boy or girl, at what age the syndrome was
diagnosed, how much secrecy there was about the diagnosis, and the degree of
open communication between the person, parents, and physicians. Management of
the condition also is related to how much freedom the individual had in
expressing feelings and behaviors. The following discussion derives from the
work of other investigators and our population study of 39 persons who had CAIS
and 18 persons who had PAIS.1
Psychiatric considerations
Secrecy,
shame, stigma
Among our respondents and
according to the findings of other investigators [14], the issues that cause
the most difficulties for individuals who have AIS are connected to secrecy,
shame, and stigma. Many complaints originate from not being informed about the
diagnosis and its implications [15]. It was believed commonly that
informing the individual as to the diagnosis would be traumatic and detrimental
to rearing [16]. Among our own sample, not a few persons complained that,
although they had undergone several routine physical examinations, they found
out about their condition on their own and then sought medical attention and clarification.
They, like others [15,17], found out about their AIS by way of a
deliberate library search, by Internet browsing, or by some chance occurrence
and were prompted to do so by features of their medical management, overheard
gossip, or “gut feelings” that aroused suspicion.
__________
1 The individuals for our study originally were recruited from AIS
support groups in the United Kingdom and the United States. Eventually, by way
of referral from clinical colleagues and a listing of our research interests on
an AIS website, others volunteered to participate. All study participants had
confirming diagnosis of the CAIS or PAIS form of the condition. Respondents
were interviewed in person and by way of an extensive written questionnaire.
The second author of this article has AIS.
Often, when CAIS was diagnosed first
at puberty, the patients were told a fabrication about faulty ovaries or
another reason why they would need to undergo surgery. It is believed now that
secrecy is detrimental and that persons who have AIS and other intersex
conditions should be informed as to their condition [18—20]. Following
diagnosis, only 51% of our respondents were told by their physician that they
had AIS. When our respondents were asked if they ever believed that it was
appropriate to not tell a person who had AIS about their condition only six
answered, “yes,” but only if the person were suicidal or specifically expressed
a desire not to know.
After finding out about their condition, most persons felt
depressed—some to the point of breakdown. Others experienced denial or shock.
After learning the truth, one of the first questions that patients ask has to
do with whether their parents had known about the condition. Secrecy is
considered a breach of trust and is harmful [20,21]. Many patients come to
realize that their parents were unaware of the condition or were told by
doctors to preserve the secret.
Only 9 of our 57 respondents (16%) were offered counseling by their
physicians. More than half of our study subjects subsequently sought out a
psychiatrist or other therapist.
Not occasionally, patients’ awareness and difficulty in dealing with
their AIS was compounded by the health care system. We, and other investigators
[22,23] found that many of our population were treated as medical oddities,
were subjected to repeated examinations, and were displayed to medical
colleagues or students or photographed without their permission. The outmoded
term “testicular female” was used frequently and insensitively in describing
their condition to them. A lack of basic knowledge of the condition was
displayed by nurses or other staff who asked women who had AIS questions, such
as “When was your last period or Pap test?”
Such encounters precipitate embarrassment and shame that is accompanied
by a perceived stigma. Kitzinger [23] wrote: “The hushed conversations, the
embarrassment of doctors, the explanations which don’t add up, lead women and
girls with AIS to the belief that they have a defect so monstrous that nobody
is willing to discuss it.”
Gender:
acceptance and rejection
Although the three Ss—secrecy,
shame, and stigma were the most frequent psychiatric concerns with which our
respondents had to deal, one of the most personal concerns is how the
individual comes to deal with the apparent disparity of being girls or women
who have an XY karyotype and testes. Persons who have AIS that live as boys or
males do not have that concern, but do have to deal with breast development,
sparse facial and body hair, and infertility.
All persons who have CAIS that were reared as girls live as women and
identify as such. Adjustment to such an assignment was reported to be
satisfactory by many investigators [24,25]. We also found that all of our
respondents who had the complete syndrome lived as women and believed that it
was the best decision for
them; however, this was not a simple solution for all. Other investigators also
found that a significant number of women expressed reservations [26].
Acceptance of assignment does not mean that assignment has been correct. It
just means that most are able to adapt and live with the handicap; however,
they might have preferred other options [27,28]. We asked our 39 subjects who
had CAIS to respond to published statements that were taken from ALIAS, an AIS
newsletter.
The first statement was: “I don’t think I am any different in feeling
than if I were born XX. .
feel very female.” Most (32 of 39; 82%)
agreed with the statement but 7 (18%) women did not.
Another statement that was presented to the respondents was: “All my
efforts over the years in presenting a female persona have left me completely
exhausted. I might just as well have had a mastectomy, cut my hair short and
lived as a celibate man. It would actually have been easier I think.”
Thirty-five (90%) subjects said that they disagreed with the statement.
When asked if they had to “work at being a woman,” 17 (44%) of the
respondents said that they “never” had to work at being a woman. The remaining
22 (56%), however, believed that at least some of the time this was
something that they had to do. Twelve of the 22 believed that they must work at
being female much of the time. The “working at” might have involved dressing in
a feminine way or using cosmetics or hair styles in a way to signal “female”
unambiguously. Some modified preferred behaviors, like altering their selection
of clothes. Not uncommonly, there were expressions of difficulty and depression
in having to overcome the knowledge that they were born XY and with testes;
however, most believed that they eventually came to terms with the knowledge of
their biology. There also were expressions of having missed typical “rites of
female passage,” such as menses and pregnancy.
Thirty-three (85%) patients in our group were content that they had been
gonadectomized; they were aware that tumor formation was a potentiality.
Reasons for discontent differed. One woman had an older sister who has AIS and
is living satisfactorily with her testes, another believed that she was more
self-confident before the surgery which only “confirmed” that there was
something “wrong” with her, and a third would have preferred watchful waiting.
A fourth believed that the resulting scars required too many difficult
explanations.
Generally, satisfaction with their situation was different among those
who had the partial condition. Those with PAIS often were ambivalent about
their assigned gender. Most (67%) believed that the gender in which they were
raised was best for them, whereas the others voiced reservations.
Most significant is the number of persons who, on their own initiative,
shifted from their gender of rearing. Among our population of 18 subjects who
had PAIS, 8 persons were raised as boys; 4 of these switched to live as woman
before our investigation. Ten members of our population who had PAIS were
raised as girls; 2 decided to live as men. Thus, 6 of 18 (33%)
individuals were assigned to a gender that they rejected later. In 5 of
these 6 individuals, the PAIS condition was confirmed by DNA or genital
fibroblast examination [29]. The sixth individual was
unaware of which confirming tests were used, but, while living as a teenage
girl, had been used in a medical school as an “example” of an individual who had
PAIS.2
We recognized four subgroups among those who had PAIS. The first two
subgroups were formed from the 10 subjects who had been raised as girls; one
group continued on to live as women (n = 8;
80%), whereas the other group had decided to live as men (n = 2; 20%). The other two subgroups were formed from the
8 subjects who were reared as boys; one subgroups consisted of the 4 who
continued to live as men and the other subgroup was made up of the 4 who
decided to live as women.
Before making the gender switch, each person evaluated many personal and
social considerations. The mean age at the shift in gender was 33.2 years
(range 18 to 46). Of the two persons who switched from living as girls to live
as men, one did so at 18 years of age and the other did it at age 30. Both of
these individuals had married as women in attempts to conform to their gender
of assignment and solidify their acceptance as women. Now, both of these men
are angry that they were brought up as girls and are particularly bothered
that, without their knowledge or informed consent, they were castrated and
subjected to vaginal reconstructive surgery and from puberty on were given
estrogens to feminize them somatically.
On the other side of the spectrum is one respondent who was raised as a
boy but who now is living as a woman. For religious and other reasons she
hesitated to change gender, although she had believed since childhood that it
would be better to live as a female. Without being consulted, from the age of
11 to 13 she had four surgical procedures to masculinize her genitalia. She was
drafted for the army, and without full medical/genetic examination, had been
told that she had KS. As an adult—still living as a man—she married a woman in
an effort to meet social expectations. Eventually, in her 40s, she developed
testicular cancer and believed that because she required genital surgery, it
would be an appropriate time to have genital reconstruction to regain her
closed vagina, get divorced, switch gender, and, henceforth, to live as a
woman. Later in life, a new clinician challenged her original diagnosis and
ordered a DNA analysis for AIS, which confirmed that she had PAIS and was
46,XY.
Another case is instructive. An individual who is now living as a woman
had been assigned as a girl at birth but was switched by a physician to live as
a boy at the age of 3 months when testes were discovered. At the age of 13, her
physician recommended that “he” have a mastectomy for his breast development.
The surgery was performed and this individual continued to live as a boy
although she had felt from early childhood that she would be more comfortable
living as a girl. At the age of 22, she felt that she could not continue to
live as a man
__________
2 Even if this person is omitted from consideration among our PAIS group, the number of those who switched from their gender of rearing is still a significant minority.
and switched to
living as a woman when she learned that she could obtain breast implants to
regain what had been removed.3
Presently, 12 (66%) of our PAIS sample are living as women and 6 (33%)
are living as men. Among these 6 are 4 men who were raised as boys. They originally
were assigned as girls but were switched by physicians to live as boys when
inguinal testes were found at the ages of 6 years, 5 years, 18 months,
and 2 weeks. Of these 4 subjects, the one who was reassigned at 18 months of
age changed to live as a woman at the age of 34. The other 3 subjects continue
to live as men. These 3 men married; 2 of them claimed that they will not shift
from their present life as married men. One acknowledged that one of his
younger siblings who also was raised as a boy did shift to live as a girl. He
acknowledged that many of his behaviors and characteristics are considered
effeminate and wishes that he could have larger male genitals to please his
partner. He writes:
“I have lived (more than 50 years) as a man now. For the last 5 years, I
get more and more doubts as to what I am really. [But] I don’t have any
thoughts about changing to a woman.” He doesn’t want to lose his wife and
adopted family and change his status.
Reports have been published about persons who had PAIS switching gender,
accepting their gender assignment, or having difficulty in establishing a
gender [28,30—32]. Preves [32] noted that the high prevalence in gender shift
among the intersexed persons that she interviewed might have been influenced by
the high educational level of her respondents. Schober [33] found that 8 of 10
subjects that she interviewed preferred being identified as intersexual, rather
than as male or female, and that 2 of the 10 were undergoing sex reassignment;
she believed that their undergoing reassignment or identifying as intersexed
might be related to their educational attainment. The education of our
respondents also might have been a factor in the gender change; all of those
who switched gender had completed college or were working on, or had attained,
a graduate degree. This also was true of those subjects in the group who had
CAIS who were most critical of their status. Obviously, general and specific
knowledge and interpretation of one’s situation, as well as concepts of opportunity,
can modify a person’s life-altering decisions.
Psychologically
relevant responses
In a standardized format, we
asked our population who had AIS, “What do you feel are the most significant
features of CAIS or PAIS with which
__________
3 This individual, while a teenager, was written up in a medical journal as an individual who had PAIS who accepted the male gender of rearing. She now claims that she never was asked searching questions about her own true gender preferences, never was informed fully of her medical condition, and never was told or believed that she had an option to change. Although this woman has remained in contact with the physician who wrote the paper more than 30 years ago, the physician has never written a follow-up publication revealing the rejected male role. This subject requested that we do not identify her in any way, including giving the citation for the publication in which she was mentioned originally.
you have to deal”? Frequent and common responses were variations of the following themes:
Secrecy, shame, and stigma
Being different in general
Concerns with infertility
Identity
How to resolve personal questions of masculinity and femininity.
We also asked our respondents “What are the most important points that need to be understood by someone with AIS”? The most common themes mentioned were:
There is more to sex and gender
than chromosomal determination; gender identity (appearance in society) is not
synonymous with sexual identity (biologic knowledge)
One who has AIS is not a freak; the condition is not something of which
to be ashamed
Being a woman or man is a mental and physical process
Typical family life with marriage and adopted children is possible
Freedom of choice is crucial in the management of one’s condition
One is not alone; support groups and the medical community can help
Lastly, is our finding that among our study population, 24 (62%) of those who had CAIS had considered suicide and 9 (23%) had attempted it. Among those who had PAIS, 11(61%) had considered suicide and 3 (17%) had attempted it. The three who had attempted suicide did so before switching from their sex of rearing. Frequently, these considerations and attempts were associated with learning of their diagnosis or a problem with a specific amorous relationship.
Kilnefelter’s
syndrome
KS is the most common sex chromosome
disorder; some studies have found a prevalence of 1 or 2 cases/1000 population
[34]. The original signal case was reported on an individual who had a 47,XXY
karyotype [35]. Since then, the syndrome has come to include several
genetic conditions in which there is an increased number of sex chromosomes.
The sex chromosome complement can vary from the most typical, 47, XXY, to XXXY,
XYY XXYY, and other combinations, and, may occur with, or independent of,
different mosaic conditions [36]. Individuals who have an XXX karyotype are
considered to have a Klinefelter’s variant by some investigators, although no Y
chromosome is present. The presence of a Y chromosome usually leads to
development as a male; persons who have XXX appear as females. Cases of individuals
who had an XXY karyotype and a female phenotype have been reported [37].
Although KS is a genetic condition, there is no evidence it is inherited or familial [38]. Most commonly, the syndrome develops from a nondisjunction during a meiotic or mitotic phase [39]. These problems occur more frequently with increased maternal age [40]. In 1997, about 10% of KS cases were detected prenatally [41]. With increasing use of amniocentesis in older, pregnant women, these numbers can be expected to increase.
Physical
features
The clinical features of KS
are extremely variable. More frequently than those who have AIS, individuals
who have KS may not be diagnosed until being worked up for infertility,
hypogonadism, or other concerns. Because many individuals who have an XXY karyotype never develop the typical symptoms of KS,
some researchers do not label someone as having KS unless prominent symptoms
develop [42].
The most consistent basic features of KS include a male phenotype with
hypogonadism, reduced or absent spermatogenesis, and increased levels of
follicle-stimulating hormone [6]. Smyth and Bremner [38] stated that the
variability in presentation is related mainly to the timing and amount of
androgen deficiency. They and others investigators [43], recommended that
school-age boys should have their testes palpated as part of a complete physical
examination; those who seem to have learning difficulties or troubles with
their peers should receive special attention.
The more supernumerary sex chromosomes that exist, the more likely are
detrimental physical and mental findings [44]. Occasionally, a finding of
cryptorchidism leads to a diagnosis of an XXY child
because the karyotype is three times more frequent in this group than in the
overall population [45].
One of the largest long-term studies of its kind, the so-called “Edinburgh study” [46], found that, at birth, babies who had KS generally were smaller in
weight, length, and head circumference than were controls. The head
circumference difference remained between the 10th and 25th percentiles and
reflected an adverse effect on brain growth [47]. A notable increase in height
velocity occurred between the ages of 5 and 8 years of age because of
the greater leg growth; however, the typical pubertal growth spurt did not
differ from controls. A tendency to central obesity was seen in 75% of
the XXY boys who were followed.
A minority of the XXY infants in the Edinburgh study was born with small
penises that prompted treatment with localized testosterone cream. These
treatments were successful in stimulating penis growth; however, by the end of puberty,
the penis was of normal size in only 77% of the boys [46]. In contrast, the
testes were normal in size at birth but failed to grow normally.
Gynecomastia in KS, is seen in from 30% to 90% of patients [44,48]. Some
reports indicate that breast development will be minimal and of little
consequence [42], whereas other investigators report that the gynecomastia will
persist [35,49]. In general, long-lasting and prominent gynecomastia does not
regress as a result of androgen treatment but might do so in less obvious cases
[6]. Gynecomastia often
is a source of great shame to the teen-age boy [50]. Reduction of
psychologically-disturbing breast growth can be accomplished by surgery or
liposuction. The incidence of breast carcinoma is significantly greater than in
the typical male [51].
Prepubertal boys who have KS tend to be taller than average with a
disproportionate leg length [52]. The onset of puberty can occur normally or be
delayed [53]. Following puberty, along with gynecomastia, there may be
diminished body and facial hair, a female pubic hair pattern, small phallus,
poor muscular development, and progressive disproportion in leg and body length.
Feminine fat distribution around the hips and abdomen is noted [38].
The presence of a normal XY cell pattern, along with KS lines, usually modifies
the expression of the syndrome so that these patients usually are diagnosed
later, display a lesser degree of gynecomastia, and show fewer feminine
characteristics and less testicular pathology [54]. Fertility becomes
increasingly doubtful as age advances; however, before infertility is
predicted, sperm analysis is required because paternity has been documented [55].
Learning
considerations
Referral to child
psychiatrists often occurs as a result of concern with schooling or behavior.
Males and females who have alterations in sex chromosome number are at
increased risk for behavioral and learning disabilities [56]. Theilgaard [48],
however, reports that the Wechsler Adult Intelligence Scale (WAIS) IQ range of
those who have XXY
or XYY is large and includes scores that
are in the normal and superior ranges.
Specific reading deficits have been reported in 50% of children who have
KS [57]. Generally, there seem to be a mild delay in language
acquisition [58] and depressed motor development [59].
Often, boys who are affected with KS are shy, passive, quiet, immature, and
dependent [59]. Typically, intelligence scores are lower than those of their
siblings and tend to reflect lower verbal skills [49]. Because many men who
have KS are never diagnosed, it is difficult to estimate or quantitatively
document the frequency or the severity of the intellectual and psychologic
problems. In addition, there seems to be considerable variability in this area;
many affected males clearly have above-average intellect [48].
Comparing XYYs and XXYs with respect to their projective test results
does not show marked differences, with the exception of sex role. “The XXY’s
have more problems with their masculine role, appear less masculine, and are
more submissive and dependent than XYY’s. In their drawings they also show less
sex-differentiation. The defensive patterns vary, the XYY’s being more evasive,
and the XXY’s showing more denial. . . [but]
the similarities far outweigh the differences” [48].
Minor deviations in motor, speech, and emotional development suggest a
common underlying pattern of altered development that may become apparent
during early childhood, before the onset of the classic physical features of
the syndrome. If
recognized, in many cases the problems can be alleviated through appropriate
intervention [43]. Such management can include specific academic schooling,
attention to speech and hearing problems, and emotional support
[43,60].
Personality
considerations
In general, XXY boys as toddlers, frequently are seen as shy and
reserved, easy to manage, and adaptable [60]. Parents often describe low levels
of activity. During school age, many are described as timid, introverted,
quiet, cooperative, eager to please, and well-liked by their teachers [61]. They
also have been described as passive, nonassertive, and not as active as their
peers [59]. In the Edinburgh study, 47% of the boys who had KS were
referred for psychiatric assistance compared with 9% of the male controls.
Teacher or parental complaints were in regard to temper tantrums, antisocial
activities, and enuresis [47]. Differences in temperament were found between
those who had XXY and those who had XYY. The former are more likely to be
bullied by others, whereas those who have XYY are more likely to be the bullies
[48].
During adolescence and transition through puberty, boys who had KS
showed low self-esteem, anger, frustration, or depression. [62]. They reported
less sexual interest in girls and had a significantly later onset of masturbation.
The number who did masturbate and the frequency of masturbation was not,
however, different between the KS and control groups [63]. High School
Personality Tests of XXY boys found them “more tender minded, sensitive,
apprehensive, self-reproaching, and insecure, while on the Bern Sex Role
Inventory they had significantly lower scores on the masculinity scale. . .less interest in girls, to date less, to have less
sexual experience, and to be less socially involved” [64]. Boys who had KS also
were likely to view themselves lower on a masculinity scale [65].
Theilgaard [48] found, after considering all aspects of overall
psychologic functioning, that the XXY and XYY groups were more alike than
different. Significant differences were noted, however, in expressed sexual
interest and activity. Those who had XYY masturbated more frequently in
childhood and adulthood, express less guilt, were younger at first intercourse,
had more partners, and reported higher libido and more unconventional sexual
activities than those who were XXY. In the “Draw A Person Test,” those who had XXY draw more differentiated images; those who had XYY
drew elaborate “overdimensionalized sexual attributes.”
The lack of masculine sense is seen from childhood in boys who have XXY but from puberty in boys who have XYY [48]. A Danish
study compared 34 men who had XXY who were
older than 20 years of age with 16 men who had XYY; it was noted that those who
were XXY had lower libido and significantly fewer had engaged in sexual
intercourse [66]. Similar findings were reported by other investigators [67].
These types of differences seem to persist. Raboch et al [68] reported that,
among men who had KS—86% of whom were married at the time—”Subjects with
Klinefelter’s syndrome date later. . .kiss
for the first time later…attempt
initial intercourse later and actually perform it later. . .Furthermore they have intercourse with a second
partner later.. .and the number of their coital partners is fewer. . .chromatin positive men belatedly fall in love for the
first time and start a long-lasting love affair at a later age.”
Gender
expression
Infrequently discussed in
medical descriptions of KS are individuals’ concerns with gender expressions
and feelings. An unknown percentage of persons who have KS experience
androgynous or feminine feelings that can develop at an early age [50]. Some
people who have KS consider themselves to be transgendered [50], others
considered themselves to be intersexed [69], and others considered themselves
to be transsexual.
One of the most noted persons who transitioned gender is Carolyn Cossey,
a “James Bond girl.” She was raised as a boy, but changed to live as a girl at
a young age, and became a famous model; her karyotype was found to be
XXXY [70].
Wyler et al [71] found that two of nine candidates that they recommended
for transsexual surgery and female hormones had KS. A host of investigators
similarly reported cases of men who had KS who transitioned to live as women or
who harbored aspects of gender dysphoria [72—74]. Walzer and Hurwitz [75] concluded
that all of the KS patients that they saw viewed their personalities as dual
male and female and wrote: “Investigators periodically report they can find no
increase in sexual deviancy in patients with a chromosomal abnormality. Only
too often the methods used to ascertain the presence of such a deviation. . .are not conducive to discovering it.” We know of two relevant
cases; one is a mathematics professor who underwent sex reassignment surgery to
live as a woman and a previous medical student who is presently in the process
of fransition. The frequency of gender change in KS is unknown.
Several papers have commented that homosexuality among those who had KS
was not found among their subjects [46] or that the prevalence was not any
different from that seen in the general population [42].
Treatment and
recommendations regarding androgen insensitivity syndrome and Klinefelter s
syndrome
Recently, KS received
attention in an attempt to derive new research priorities and stimulate
investigation in this area of intersexuality [76]. New reviews of this
condition, along with focus on AIS, are welcome.
AIS and KS benefit from many similar treatments and recommendations.
Children and adolescents who have intersex conditions deserve, and benefit
from, appropriate psychiatric care and counseling. The diagnosing physician
should encourage such attention, even if it is not requested by the patient or
parent. Therapy should be age-appropriate, honest, and open with sensitivity to
intersex issues. Psychiatric care in an empathic setting is preferable to
concealment or self-discovery
in an environment that is devoid of support [77]; support should be ongoing as
necessary.
Goodall [78] advised that children do not view their troubles as adults
do; attempts to protect them from adult knowledge may leave them vulnerable to
a shocking revelation at exactly the age when conformity with peers and sexual
identity are important and also could breed noncompliance with treatment. “A
better approach is to unfold the truth state by stage, matching simple
statements to the child’s conceptual growth until the personal implications are
finally realized as part of a maturing process.” [78]. Bock [42] reminds us
that “when the truth is withheld, children often suspect that their parents are
hiding something and may imagine a condition that is worse than their actual
diagnosis.”
Simultaneous with informing the patient or parents about the diagnosis
and discussing medical concerns, there should be adequate opportunity for
questions and debate. Contact with a particularly aware individual who has a
similar condition or to a support group is advocated.4 Those who have AIS or KS
and their parents have found such associations to be helpful.
Parental counseling can start as soon as the child’s condition is
determined and can precede counseling of the child [27,79,80]. The type and
magnitude of this assistance depends on the individual child, the specific
condition, and the parent’s own resources—psychologically, socially, and
otherwise [34].
Specific aspects of psychotherapy that need attention are the items that
were identified above in the AIS section; these are applicable to patients who
have KS. Counseling should, in addition, cover concerns with body appearance
and physiology, relationships among peers and potential sexual partners [81],
genetics [82], and sexual functioning.
Those who have AIS or KS and live as girls or women have to reconcile
that they are living with an anomalous karyotype, are born with testes, will
not menstruate, and will be infertile; they will miss many of the social
milestones of the typical female. Those who have AIS or KS and live as boys or
men need to deal with breast development and other physical differences from
their peers and the knowledge that they probably will be infertile. Although
most boys and girls have no difficulty in resolving questions of gender, all
have to deal with associated considerations. Some will choose to live in a
gender other than that in which they were reared; generally, this will not be
easy. Manifestations of effeminacy or masculinity in contrast with their chosen
gender is a topic that often will need to be addressed. Teenage boys and girls
are particularly sensitive about how their peers perceive them and how well
they integrate with them.
For those who are dealing with gender issues, Hoffman [83] advised that
therapists assist the patient to challenge disturbing stereotypes of
masculinity and
__________
4 When not convenient geographically, support groups are available by way of the Internet. Some AIS groups limit themselves to those who live as women although a few groups welcome men as well. Some groups are led by parents and others are directed by intersexed individuals.
femininity and
to explore how the individual learned to be a boy or girl. It also is helpful
to offer basic information about genital developmental processes and
distinguishing sex from gender [84] and to keep in mind that concepts of gender
are culturally based [85].
If gender change is considered
appropriate, support is crucial.
Among those persons who have AIS and live as women, vaginal enlargement
is an issue that often needs to be addressed. Surgical intervention has been
challenged, particularly when it is done without the informed consent of the
patient [86]. Gooren [87] wrote: “Dilation is the intervention of first choice.
It is self- performed, using a progressively enlarged series of penis-shaped
dilators. .
.Gentle pressure is applied into the
vaginal outlet, and over the course of several weeks or longer a 10-minute
period of dilation twice a day may suffice. Psychological counseling is needed
during this period as there may be a phobia of vaginal penetration or the girls
may equate the procedure with masturbation, particularly when the dilator is
obtained from a nonclinical source.” Moen [88] also found that self-dilation
and cautious intercourse was effective in enlarging the vagina. Parents should
be consulted and counseled about these practices because they might have their
own questions and concerns regarding its timing and other ramifications.
Appropriate counseling regarding vaginal functioning should be ongoing [31].
Advice regarding testosterone or dihydrotestosterone (DHT)
administration to those who have AIS and live as men, as well as those who have
KS, varies and is based on little experimental data. The value of androgens for
those who have AIS probably is related to the exact nature of the mutation that
is involved [89] because it helps a few people but not most. In large dosages,
testosterone was successful in producing significant phallic growth and
enhancing male living [90]. DHT was helpful in restoring male genital
development in an infant who had PAIS [91]. For individuals who have been
gonadectomized, as well as those who have KS, hormonal management helps to
prevent osteoporosis and other medical conditions.
For undermasculinized boys or men who have KS, Nielsen and colleagues
[66,92] and Forest [4] recommended testosterone treatment that was started
early in puberty. They claimed that it helped to prevent the development of
deviations in behavior and learning abilities at school. Testosterone also was
noted to stimulate and increase general activity and well-being. Treatment of
KS with testosterone is suggested, beginning at 11 to 12 years in accord with
the patient’s state of well-being, degree of virilization, and growth [44]. In
general, parenteral androgens were more effective in inducing virilization and
were safer than oral preparations [6].
How individuals respond to their intersex conditions varies greatly.
From the varieties of findings in the accumulated reports, it should not be
anticipated that a person who has AIS or KS will fit any model or demonstrate
any particular personality characteristics. Stereotyping those who have AIS or
KS into homogeneous profiles should be resisted. As in other aspects of medicine,
children and adolescents need to be dealt with as individuals.
References
[1] Uehara 5, Tamura M, Nata M,
Kanetaki J, Hashiyada M, Terada Y. Complete androgen insensitivity in a 47,XXY
patient with uniparental disomy for the X chromosome. Am J Med Genet
1999;86:107—l1.
[2] Quigley CA, De Bellis A, Merschke KB, El-Awady MK, Wilson EM, French FS.
Androgen receptor defects: historical, clinical and molecular perspectives.
Endocr Rev 1995;16(3):
271 321.
[3] Gottlieb B, Pinsky L, Beitel LK, Trifiro M. Androgen insensitivity. Am J
Med Genet 1999; 89(4):210—7.
[4] Forest MG. Diagnosis and treatment of disorders of sexual development. In:
DeGroot U, Jameson JL, editors. Endocrinology, vol. 3. 4th edition. Philadelphia: W. B. Saunders Company; 2001. p. 1974—2010.
[5] Imperato-McGinley J, Zhu Y-S. Gender and behavior in
subjects with genetic defects in male
sexual differentiation. In: Pfaff OW, Arthur PA, Etgen AM, Fahrbach SE, Rubin
RT, editors. Hormones, brain and behavior, vol. 5. San Diego (CA):
Academic Press; 2002. p.
303—46.
[6] Grumbach MM,
Huges IA, Conte FA.
Disorders of sex differentiation. In: Kroncnberg HM, Melmed S, Polonsky
KS, editors. Williams textbook of endocrinology. Philadelphia: W. B.
Saunders;
2003. p. 842—1002.
[7] Gottlieb B, Beitel LK, Lumbroso R, Pinsky L, Trifiro M. Update of the
androgen receptor gene mutations database. Hum Mutat 1999; 14(2): 103—14.
[8] Batch JA, Patterson MN, Hughes LA. Androgen insensitivity syndrome. Reprod
Med Rev 1992;1:131—50.
[9] McEwen BS. Gonadal steroid influences
on brain development and sexual differentiation. In: Greep RO, editor.
Reproductive physiology IV, international review of physiology, vol. 27.
Baltimore (MD): University Park Press; 1983. p. 99—145.
[10] Hines M. Sexual differentiation of human brain and behavior. In: Pfaff DW,
Arthur PA, Etgen AM, Fahrbach SE, Rubin RT, editors. Hormones, brain and
behavior, vol. 4. San Diego (CA):
Academic Press; 2002. p. 425—62.
[11] Quigley CA, Friedman KJ, Johnson A, Lafreniere RG, Silverman LM, Lubahn
DB, et al. Complete deletion of the androgen receptor gene: definition of the
null phenotype of the androgen insensitivity syndrome and determination of
carrier status. J Clin Endocrinol Metab 1992;
74(4):927—33.
[12] Evans BAJ. Phenotypic diversity in siblings with partial androgen
insensitivity syndrome. Arch Dis Child l997;76:529—31.
[13] Viner RM, Tech Y, Brown BD, Patterson MN, Hughes IA. Androgen
insensitivity syndrome:
a survey of diagnostic procedures and management in the UK. Arch Dis Child 1997;77(4):305 —9.
[14] Kessler Si. Lessons from the intersexed. New Brunswick (NJ): Rutgers University Press; 1998.
[15] Anonymous. Once a dark secret. Br Med J l994;308:542.
[16] Natarajan A. Medical ethics and truth-telling in the case of androgen
insensitivity syndrome. Can Med Assoc J 1996;l54:568 70.
[17] Groveman SA. The Hanukkah bush: ethical implications in the clinical
management of intersex. J Clin Ethics 1998;9(4):356—9.
[18] Kemp BD, Groveman SA, Anonymous, Tylo HD, Irwin KM, Natarajan A, et al.
Sex, lies and androgen insensitivity syndrome. Can Med Assoc J 1996; 154(12):
1829—33.
[19] Warne GL. The baby of uncertain sex. Pediatr Surg 1992;7:244—8.
[20] Diamond M. Pediatric management of ambiguous and traumatized genitalia. J
Urol l999;162:
1021—8.
[21] Liao LM. Learning to assist women born with atypical genitalia: joumey
through ignorance, taboo and dilemma. J Reprod Infant Psychol 2003;21(3):229—38.
[22] Wilson BE. Androgen insensitivity syndrome. Available at:
www.emedicine.comfPED/ topic2222.htm. Accessed March 4, 2003.
[23] Kitzinger C.
Women with androgen insensitivity syndrome. In: Ussher JM, editor. Women’s
health. Leicester (UK): British Psychological Society; 2000. P. 387—94.
[24] Wisniewski AB, Migeon CJ, Meyer-Bahlburg HF, Gearhart JP, Berkovitz GD,
Brown TR, et al. Complete androgen insensitivity syndrome: long-term medical,
surgical, and psychosexual outcome. J Clin Endocrinol Metab 2000;85(8):2664 9.
[25] Hines M, Ahmed SF, Hughes IA. Psychological outcome and gender-related
development in complete androgen insensitivity syndrome. Arch Sex Behav
2003;32(2):93 —101.
[26] Migeon CJ, Wisniewski AB, Gearhart JP, Meyer-Bahlburg HFL, Rock JA, Brown
TR, et al. Ambiguous genitalia with perineoscrotal hypospadias in 46,XY
Individuals: long-term medical, surgical, and psychosexual outcome. Pediatrics
2002; 11 0(3):61 6—21.
[27] Diamond M, Sigmundson HK. Management of intersexuality: guidelines for
dealing with persons with ambiguous genitalia. Arch Pediatr Adolesc Med
1997;151:1046—50.
[28] Kuhnle U, Krahl W. The impact of culture on sex assignment and gender
development and gender development in intersex patients. Perspect Biol Med
2002;45(1):85 103.
[29] Edelstein RA, Carr MC, Caesar R, Young M, Atala A, Freeman MR. Detection
of human rogen receptor mRNA expression abnormalities by competitive PCR. DNA
Cell Biol 1994; l3(3):265 73.
[30] Gooren LJG, Cohen-Kettenis PT. Development of male gender identity/role
and a sexual orientation towards women in a 46,XY subject with an incomplete
form of the androgen insensitivity syndrome. Arch Sex Behav 199l;20(5):459—70.
[31] Vates TS, Fleming P, Leleszi JP, Barthold JS, Gonzalez R, Peclmutter AD.
Functional, social, and psychological adjustment after vaginal reconstruction.
J Urol 1999; 162:182—7.
[32] Preves SE. Negotiating the constraints of gender binarism: intersexuals’
challenge to gender categorization. Cuff Soc 2000;48(3):27 50.
[33] Schober JM. Sexual behaviors, sexual orientation and gender identity in
adult intersexuals: a pilot study. J Urol 2001;165(6 part 2):2350 3.
[34] Nielsen J, Wohlert M. Sex chromosome abnormalities found among 34,910
newbom children:
results from a 1 3-year incidence study in Arhus, Denmark. Birth Defects Orig
Artic Ser 1991; 26:209—23.
[35] Kleinfelter HRJ, Reifenstein ECJ, Albright F. Syndrome characterized by
gynecomastia, aspermatogenesis without a-Leydism, and increased excretion of
follicle-stimulating hormone. J Clin Endocrinol 1942;2:6l5—27.
[36] Mandoki MW, Sumner GS, Hoffman RP, Riconda DL. A review of Klinefelters
syndrome in children and adolescents. J Am Acad Child Adolesc Psychiatry 1991
;30(2): 167 72.
[37] Schmid M, Guttenbach M, Endres H, Terruhn V. A 47,XXY female with unusual
genitalia. Hum Genet 1992;90:346—9.
[38] Smyth C, Bremner WJ. Klinefelter syndrome. Arch Intern Med l998;158:1309
14.
[39] Abruzzo M, Hassold TJ. Etiology of nondisjunction in humans. Environ Mol
Mutagen 1995; 25(Suppl 26):38—47.
[40]
Carotheres AD, Fillippi G. Klinefelter’s syndrome in Sardinia and
Scotland: comparative studies of parental age and other aetiological
factors in 47,XXY. Hum
Genet 1 988;8 1:71 —5.
[41] Abramsky L, Chapple J. 47,XXY (Klinefelter syndrome) and 47,XYY: estimated
rates of and indication for postnatal diagnosis with implications for prenatal
counseling. Prenatal Diagnoses
1997;17:363—8.
[42] Bock R. A guide for XXY males and their families. Available at:
http://www.nichd.nih.gov/ publications/pubs/klinefelter.htm. Accessed August
11, 2003.
[43] Battin J, Malpuech G, Nivelon JL, Garandeau P, Freycon F, Sultan C, et al.
Le syndrome de Klinefelter en 1993. Resultats d’une enquete multicentrique sur
cinquante-huit cas et revue de Ia literature. [Klinefelter syndrome in 1993.
Results of a multicenter study on 58 cases and review of the literature]. Ann
Pediatr (Paris) I 993;40(7):432 7 [in French].
[44] Chen H. Klinefelter syndrome. Available at:
http://www.emedicine.com/PED/topic1252.htm. Accessed August II, 2003
[45] Topper E,
Doickerman Z, Prager-Lewin R, Kaufman H, Maimon Z, Laron Z. Puberty in 24
patients with Klinefelter syndrome. Eur J Pediatr 1982;139:8— 12.
[46] Ratcliffe S. Long term outcome in children of sex chromosome
abnormalities. Arch Dis Child
1999;80(2): 192—5.
[47] Ratcliffe SG, Masera N, Pan H, Mckie M. Head circumference and IQ of
children with sex chromosome abnormalities. Dev Med Child Neurol Suppl
1994;36:533 44.
[48] Theilgaard A. A psychological study of the personalities of XYY and XXY
men: assessment and discussion. Acta Psychiatr Scand 1 984;69(Suppl 31 5):70
124.
[49] Ratcliffe 5, Bancroft J, Axworthy D, McLaren W. Klinefelter’s syndrome in
adolescence. Arch Dis Child 1982;57:6 12.
[50] McKinlay 1W. The KS Story: You are not alone. Peebles, Scotland, in press.
[51] Hultbom R, Hanson C, Kopf I, Verbiene I, Wamharmmas R, Weimarck A.
Prevalence of Klinefelter’s syndrome in male breast cancer patients. Anticancer
Res 1997;17:4293—7.
[52] Schibler D, Brook CG, Kind HP, Zachmann M, Prader A. Growth and body
proportions in 54 boys and men with Klinefelter’s syndrome. Helv Paediatr Acta
1974;29:325—33.
[53] Berenguer B, de Ia Cruz L, de Ia Plaza R. The role of lipoaspiration in
defeminization of Klinefelter syndrome: a case report. Ann Plast Surg 1999;43:306—8.
[54] Theilgaard A. A psychological study of the personalities of XYY and XXY
men. Acta Psychiatr Scand 1984;69(Suppl 315):14—5.
[55] Laron Z, Dickman Z, Zamir R, Galatzer A. Paternity in Klinefelter’s
syndrome—a case report. Arch Androl 1982;8(2):149—51.
[56] Geschwind DH, Boone KB, Miller BL, Swerdloff RS. Neurobehavioral phenotype
of Klinefelter syndrome. Ment Retard Dev Disabil Res Rev 2000;6: 107 16.
[57] Graham J, Bashir A, Stark R, Silbert A, Walzer S. Oral and written
language abilities of XXY boys: Implications for anticipatory guidance.
Pediatrics 1 988;8 1:795—806.
[58] Rovet J, Netley C, Keenan M, Bailey J, Stewart D. The psychoeducational
profile of boys with Klinefelter syndrome. J Learn Disab I 996;29: 180 96.
[59] Nielsen J, Sorensen AM, Sorensen K. Follow-up until age 7 to 11 of 25
children with sex chromosome abnormalities. Birth Defects Orig Artic Ser I
982;1 8(4):61 —97.
[60] Walzer 5, Bashir AS, Silbert AR. Cognitive and behavioral factors in the
learning disabilities of 47, XXY and 47, XYY boys. Birth Defects Orig Artic Ser
1991;26(4):45 58.
[61] Walzer 5, Bashir Jr A, Graham Jr JM, Silbert AR, Lange NT, DeNapoli MF, et
al. Behavioral development of boys with X chromosome aneuploidy: Impact of
reactive style on the educational interventions for learning deficits. Birth
Defects Orig Artic Ser 1986;22: 1—21.
[62] Bender BG, Harmon R, Linden M. Psychosocial adaptation of 39 adolescents
with sex chromosome abnormalities. Pediatrics 1 995;96:302—8.
[63] Bancroft J, Axworth D, Ratcliffe S. The personality and psycho-sexual
development of boys with 47 XXY chromosome constitution. J Child Psychol
Psychiatry 1982;23: 169 80.
[64] Roy A. Psychiatric disorders in relation to Klinefelter’s syndrome. In:
Bandmann E-J, Breit R, Perwein E, editors. Klinefelter’s syndrome. Berlin: Springer-Verlag; 1984. p. 192—201.
[65] Martinius J. Psychiatric aspects of Klinefelter’s syndrome in adolescence.
Klinefelter’s syndrome. Heidelberg, Germany: Springer-Verlag; 1984.
[66] Nielsen J, Johnsen 5, Sorensen K. Follow-up 10 years later of 34
Klinefelter males with karyotype 47,XXY and 16 hypogonadal males with karyotype
46 XY. Psychol Med 1980;10:
345 —52.
[67] Sorensen K. Physical and mental development of adolescent males with
Klinefelter syndrome. Harm Res 1992;37(Suppl 3):55—61.
[68] Raboch J, Mellan J, Starka I. Klinefelter’s syndrome: sexual development
and activity. Arch Sex Behav 1979;8:333—9.
[69] Butler J. Groom’s intersex quandary. The West Australian Saturday 2002;5.
[70] Cossey C. My story. London: Faber and Faber; 1991.
[71] Wyler J, Battegay R, Krupp 5, Rist M, Rauchfleisch U. Der transsexualismus
und dessen
Therapie.
Transsexualism and its therapy. Schweiz Arch Neurol Neurochir Psychiatr 1979;
124(1):43—58 [in German].
[72] Baker HJ, Stoller RJ. Can a biological force contribute to gender
identity? Am J Psychiatry 1968;124(l2):1653—8.
[73] Seifert D, Windgassen K. Transsexual development of a patient with
Klinefelter’s syndrome. Psychopathology 1995;28(6):312—6.
[74] Parks JS. Cognitive style and gender role in persons with sex chromosome
aberrations. Hosp Pract 1977;12(l0):93—102, 107—8.
[75] Walzer S, Hurwitz I. Psychosexual ambiguity in Klinefelter’s syndrome.
Semin Psychiatry 1970; 2(l):53—64.
[76] Simpson JL, de Ia Cruz F, Swerdloff RS, Samango-Sprouse CS, Kakkebaek
NE, Graham JMJ, et al. Klinefelter syndrome: expanding the phenotype and
identifying new research directions. Genet Med 2003;5:460—8.
[77] Pinsky L, Trifiro M. Androgen insensitivity syndrome. Gene reviews.
Available at: http://www.
geneclinics.org/servlet/access?db=geneclinics&site=gt&id=8888891
&key=6jQCCNiJukpy4&gry&fcn=y&fw=gC1J
&filename=/profiles/androgen/index.html. Accessed on March 27, 2004.
[78] Goodall J. Helping a child to understand her own testicular feminization.
The Lancet 1991; 337(8732):33 5.
[79] McGillivray BC. Genetic aspects of ambiguous genitalia. Pediatr Clin North
Am 1992;39(2):
307—17.
[80] Slijper FME, Drop SLS, Molenaar JC, Scholtmeijer RJ. Neonates with
abnormal genital development assigned the female sex: parent counseling. J Sex
Educ Ther 1994;20(l):9— 17.
[81] Efthim PW, Kenny ME, Mahalik JR. Gender role stress in relation to shame,
guilt, and externalization. J Couns Dcv 2001;79:430—8.
[82] Drop SL, Boehmer AL, Slijper FM, Nijman JM, Flazebroek FW, Niermeijer MF.
Differential diagnosis and treatment of girls with 46XY-karyotype and androgen
insensitivity syndrome. Ned Milit Geneeskd Tijdschr 2001;145(14):665 9.
[83] Hofffiian RM. The measurement of masculinity and femininity: historical
perspective and implications for counseling. J Couns Dev 2001 ;79:472 85.
[84] Diamond M. Sex and gender are different: sexual identity and gender
identity are different. Clin Child Psychol Psychiatry 2002;7(3):320—34.
[85] Kuhnle U, Krahl W. The impact of culture on sex assignment and gender
development in intersex patients. Perspect Biol Med 2002;45(1):85 103.
[86] Beh HG, Diamond M. An emerging ethical and medical dilemma: should
physicians perform sex assignment surgery on infants with ambiguous genitalia? Mich J Gend Law 2000;7(1): 1—63.
[87] Gooren LJG. Androgen-resistance syndromes: considerations of gender
assignment. In: Bardin CW, editor. Current therapy in endocrinology and
metabolism. 6th edition. St. Louis (MO):
Mosby; 1997. p. 380—4.
[88] Moen MH. Creation of a vagina by repeated coital dilatation in four
teenagers with vaginal agenesis. Acta Obstet Gynecol Scand 2000;79(2): 149—50.
[89] Weidermann W, Peters B, Romalo G, Spindler K-D, Schweikert H-U. Response
to androgen treatment in a patient with partial androgen insensitivity and a
mutation in the deoxyribonucleic acid-binding domain of the androgen receptor.
J Clin Endocrinol Metab 1 998;83(4): 1173—6.
[90] Grino PB, Isidro-Gutierrez RF, Griffin JE, Wilson JD. Androgen resistance
associated with a qualitative abnormality of the androgen receptor and
responsive to high-dose androgen therapy. J Clin Endocrinol Metab 1989;68:578—84.
[91] Ong YC, Wong HB, Adaikan G, Yong EL. Directed pharmacological therapy of
ambiguous genitalia due to an androgen receptor mutation. The Lancet
1999;354:1444—5.
[92] Nielsen J, Pelsen B, Sorensen K. Follow-up of 30 Klinefelter males treated
with testosterone. Clin Genet 1988;33:262—9.